Introduction to Pharmacokinetics
Pharmacokinetics is a comparatively new science that started to gain prominence in the late 1950's and 1960's, borrowing many of its concepts and approaches from the engineering field and with a significant emphasis on mathematics.
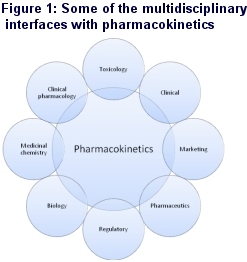
Now, however, it has evolved into a much more physiologically and pharmacologically based science, impacting on almost every stage of the drug discovery and development process and interfacing with many different discovery and development functions within the pharmaceutical / biotechnology industry and with regulatory authorities such as the FDA, EMEA and the MHLW (Figure 1).
What is Pharmacokinetics?
So what is pharmacokinetics? I define it very simply as 'what the body does to the drug' and it should always be considered in context to another scientific discipline, pharmacodynamics, or 'what the drug does to the body'.
Effectively pharmacokinetics is used as a surrogate for pharmacodynamics, the main interest of drug developers, since this results in the efficacy of a drug where the response is desired or the side effects (toxicity) where undesired.
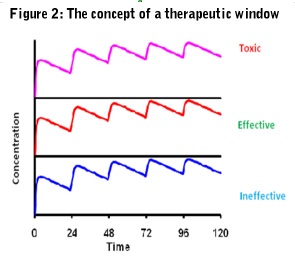 Ideally, the pharmacokinetics of a drug should always be related to its pharmacodynamics so therapeutic and side effect thresholds can be established and a therapeutic window defined (Figure 2).
Ideally, the pharmacokinetics of a drug should always be related to its pharmacodynamics so therapeutic and side effect thresholds can be established and a therapeutic window defined (Figure 2).
Pharmacokinetics can then be used to design dosage regimens and adjust them in event of drug-drug interactions and special patient groups. To use this most effectively, however, it is important to understand the basic mechanisms that govern the pharmacokinetics of a drug.
Fundamentals of Pharmacokinetics
Pharmacokinetics can be described by the following three basic processes:
Absorption
Defined as the transfer of a drug from the site of administration to the site of measurement. This should not be confused with bioavailability, which is defined as the fraction of intact drug, or active moiety if a prodrug, reaching the systemic circulation. It is quite possible to have an oral drug that is 100% absorbed but with 0% bioavailability because, for example, due to high hepatic first pass metabolism.
Distribution
Defined as the reversible transfer of a drug to and from the site of measurement. In the majority of cases the concentration of a drug is measured in plasma, serum or blood but often the site of action is within other tissues. It is thus important to understand the factors governing the transfer of the drug to the desired site of action.
Elimination
Defined as the irreversible transfer of a drug from the site of measurement and includes drug metabolism, renal excretion, biliary excretion as well as several other minor routes. Drug metabolism is included in elimination since, if a drug is chemically modified, it is no longer the original compound and, unless the metabolism is reversible, has been irreversibly removed from the site of measurement. It is important, therefore, that any bioanalysis of the drug is specific if a true understanding of the pharmacokinetics of a drug is to be obtained. If information is required on a metabolite because it is pharmacologically or toxicologically active, it should be studied separately, since invariably non-specific assays will lead to erroneous conclusions.
The drug Discovery and Development Process
Drug discovery and development is a risky business since it has been estimated that of every 5000 compounds synthesized only 5 will reach clinical trials and of those that do enter clinical development only 5-20% (depending on therapeutic area) will actually get to the market taking about 14 years of research and development at an average cost of approximately $1 billion.
Many drugs fail after entering the development process and obviously, the costs become far greater the later in the development process the drug fails.
Another disturbing fact is that only 3 of every 10 drugs marketed will ever recover their research and development costs, especially since with intense competition, exclusivity has declined from several years to just a few months. Thus, for a drug to be really successful with a good return on investment it necessary not only to gain regulatory approval but also to demonstrate significant superiority over the competition at the time the drug enters the market.
One of the most effective ways of achieving this is to have a clear idea of the desired profile of the drug right at the beginning of the project, so a screening funnel can be designed from the start to ensure that the desired attributes, including the pharmacokinetic attributes, are built into the molecule.
The Impact of Pharmacokinetics
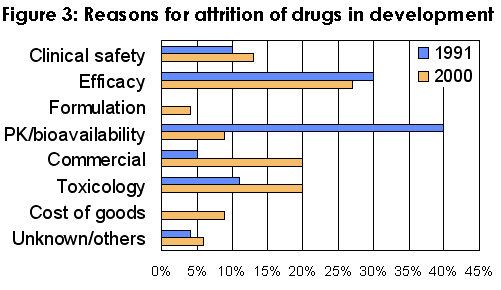
Analysis has shown that inappropriate pharmacokinetics was a major contributor to attrition of drugs in the 1990's although it had dramatically improved by early 2000 (Figure 3).
This was accomplished by a deliberate policy of 'front loading' pharmacokinetic considerations into the drug discovery process by introducing a variety of screens, many being in vitro assays, designed to help select compounds with desirable pharmacokinetic properties.
In addition, in 1997 Lipinski introduced the 'rule of five' into medicinal chemistry (cLog P <5; molecular mass <500; number of hydrogen bond donors <5; number of hydrogen bond acceptors <10) which has further driven an awareness of factors needed to produce compounds with more drug-like properties.
No Time for Complacency
Well, one might be excused for thinking that with all these activities we have the pharmacokinetic optimization of drugs mastered, but unfortunately not. The introduction of these assays has proven to be essential to prevent wasted resource on pharmacokinetically inappropriate compounds, as well as understanding absorption, distribution and clearance mechanisms that can enable the most effective use of a drug. However, the great temptation is still to view each one of the pharmacokinetic properties in isolation, often setting up the screening funnel as a series of individual filters, each getting finer and finer as compounds progress down the funnel.
This can prove disadvantageous, since concentrating on and optimizing just one pharmacokinetic property can completely negate another. Also, with the concept of a series of filters, it can become too strict, with some very acceptable compounds destined for the trash can.
The problem is that, one can become deluded into thinking that one is very successful, since attrition appears to decrease, the rejected compounds are forgotten and, unfortunately, the pipeline dries up.
This has been and is an all too common problem for the industry and a retrospective analysis of successful marketed compounds versus different companies' criteria for entering development, reveals that many drugs would not have made it in the current environment and valuable medicines would have been lost to the patient.
Looking to the Future
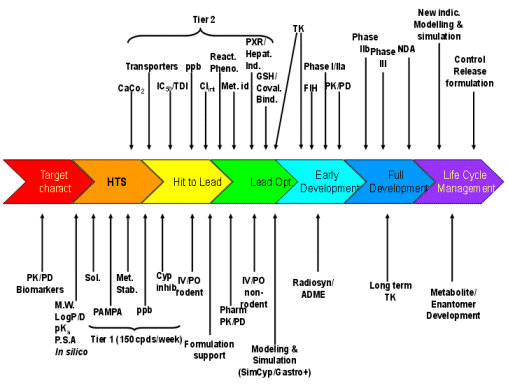
Can we do better? A rhetorical question I know, because I firmly believe we can by the application of multidimensional integration of non-clinical pharmacokinetic data to determine the overall risk of failure in later development. The assays and their typical entry points in the drug project life cycle are shown in Figure 4 below.
All the data are combined, taking into account the effect of one parameter upon another, bringing together in vitro and in vivo data with modeling and simulation to predict the situation and the variability in the patient.
Whilst this may increase the number of compounds entering early clinical development, cost and resource can be minimized by using an exploratory development approach with highly focused studies, such as microdosing, to reach key quality decisions as quickly as possible.
This will not only help accelerate the discovery and early development of a candidate drug, leading to proof of concept in man, but also provides much more robust data for key decision making with less chance of prematurely discarding potentially important new medicines.
Figure 4: Typical entry points of pharmacokinetic evaluations through a project life cycle
Legend to Figure 4:
Target charact.: Characterization and validation of the target including both pharmacological and toxicological aspects | IV/PO non-rodent: Absolute bioavailability study in a non-rodent species usually selected for toxicology, also determining clearance, volume & half-life |
HTS: High throughput screening | Radiosyn/ADME: Synthesis of radiolabelled compound with typical ADME studies such as excretion/balance studies in the toxicology species and human: tissue distribution (quantitative whole body autoradiography): metabolite identification in the toxicological species and human |
Lead Opt.: Lead optimization | TK: Toxicokinetics and assumes validation of bioanalytical method if under GLP |
PK/PD Biomarkers: Pharmacokinetic/pharmacodynamic correlations using pharmacological models & biomarkers if available | CaCo2: Permeability using a CaCo2 cell monolayer (other cell lines can be used such as MDCK) also gaining information on transporters (primarily pgp) from the ratio of drug moving apical→basal to basal→apical sides |
M.W. : Molecular weight | Transporters:In vitro assays using cells transvected with a specific transporter |
P.S.A.: Polar surface area | IC50/TDI: Determination of the IC50 for competitive inhibition of different cytochromes and determination of time dependent or mechanism based inhibition, primarily CYP 3A4 |
In silico: Computer predicted calculations of pharmacokinetic attributes | Clint: Intrinsic clearance determinations typically using microsomes and/or hepatocytes of the toxicological species and human. |
Sol: Solubility | React. Pheno.: Reaction phenotyping determining the extent of elimination by each metabolic route including those of individual cytochromes. |
PAMPA: Permeability assay using artificial membranes | Met. i.d.: Metabolite identification, both in vitro and in vivo |
Met. Stab: Metabolic stability typically using rodent & human microsomes | PXR/Hepat. Ind.: Determination of induction potential initially by binding to the nuclear receptor, PXR, of the toxicological species and human, followed by respective functional hepatocyte assays |
ppb: Plasma protein binding | GSH/Coval. Bind.: Determination of potential for reactive metabolite formation, initially by reduced glutathione trapping, if necessary followed by covalent binding typically using radiolabelled compound and exhaustive extraction following incubation with microsomes. |
Cyp inhib.: Inhibition of major expressed human cytochromes (e.g. CYP 1A2, 2C9, 2D6, 2E1, 3A4) | FIH: First in human studies and assumes a sensitive, robust and validated bioanalytical method |
IV/PO rodent: Absolute bioavailability study in a rodent species also determining clearance, volume & half-life. Assumes development of a robust bioanalytical method | PK/PD: Pharmacokinetic/pharmacodynamic correlations using biomarker, pharmacological and/or efficacy measures. Later phases of clinical development will use population approaches |
Pharm. PK/PD: Pharmacokinetic support for pharmacology studies with pharmacokinetic /pharmacodynamic correlations |
|